01 gennaio 2022
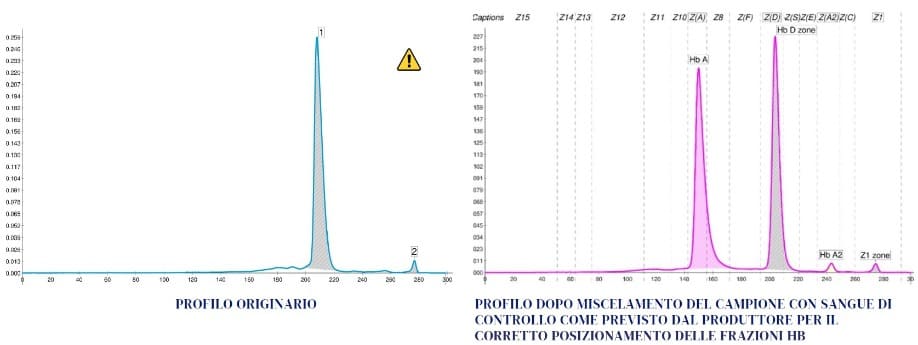
INTERPRETAZIONI
IPOTESI
Quando l’Hb A risulta assente, dobbiamo sempre allarmarci e capire in tempi, possibilmente brevi, come può essersi prodotta tale condizione. È vero anche che nei soggetti non trasfusi e con Hb A assente è comunque sempre presente un meccanismo più o meno efficace di compenso al difetto Hb. Nei casi di talassemia major, ad esempio, in mancanza di catene β, è l’Hb F che in parte assolve a tale compito di compenso.
Il caso qui documentato è molto diverso dal solito e ci induce ad alcune considerazioni:
In entrambi i quadri i restanti geni si devono supporre non funzionanti (talassemici).
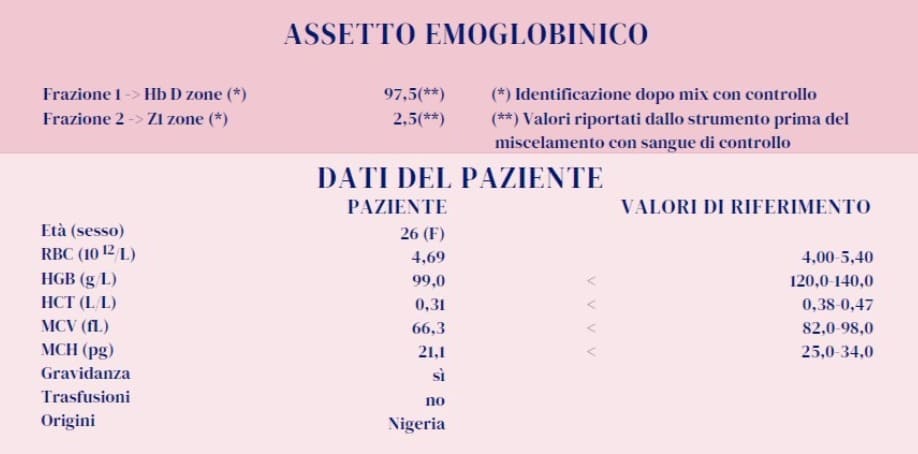
La condizione omozigote risulta dal fatto che questa donna ha ereditato da entrambi i genitori la variante G-Philadelphia con in cis un gene α deleto. Tali risultati confermano le ipotesi iniziali. L’Hb G-Philadelphia costituisce la stragrande maggioranza dell’emoglobina prodotta (97,5%) e sopperisce in modo efficace alla mancanza di Hb A. Il picco in «zona Z1» (2,5%) rappresenta l’Hb A2 con le catene α mutate. La variante Hb G-Philadelphia è stata descritta soprattutto in Africa, negli Stati Uniti e anche in Italia, in molti casi associata in vario modo all’alfa talassemia (1-3).
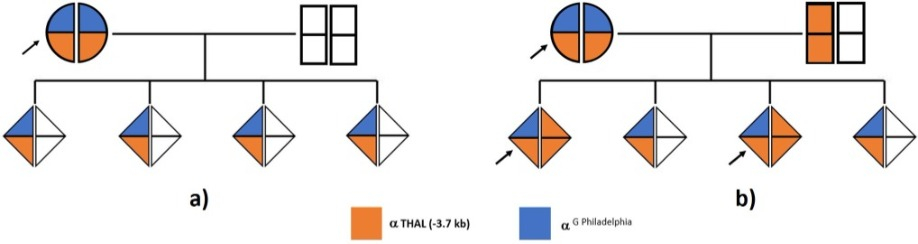
CONCLUSIONI
I figli della probanda saranno tutti portatori obbligati del doppio difetto G-Philadelphia/-3.7kb su un solo allele se, e solo se, il padre, partner della paziente, possiede geni alfa normali (Fig. a). Può essere interessante osservare che se il partner fosse portatore di due geni alfa deleti sullo stesso allele (es. alfa --MED), il 50 % dei figli potrebbe avere una malattia da Hb H associata alla Hb G-Philadelphia allo stato eterozigote (Fig. b). In tal caso ne risulterebbe un assetto Hb molto simile a quello della madre ma con la presenza di un picco in «Z15 zone» (Hb H) e Hb A2 molto ridotta; l’anemia con ogni probabilità risulterebbe più importante (vedere Caso clinico 3). Una consulenza genetica appropriata dovrà quindi basarsi sulla caratterizzazione dei difetti globinici presenti nella probanda e nel partner (3,4).
BIBLIOGRAFIA
1.Sciarratta GV, Sansone G, Ivaldi G, Felice AE, Huisman TH. Alternate organization of alpha G-Philadelphia globin genes among U.S. black and Italian Caucasian heterozygotes. Hemoglobin 1984;8(6):537-47
2.Pardoll DM, Charaohe S, Hjelle BL, Jones R, Phillips JA, et al. Homozygous α Thalassemia/HB G Philadelphia. Hemoglobin 1982;6(5):503-15.
3.HbVar database for human hemoglobin variants and thalassemia mutations. http://globin.bx.psu.edu/hbvar/menu.html bx.psu.edu (ultimo accesso 29.10.2021).
4.Barberio G, & Ivaldi G. (2020). Emoglobinopatie. Dalla diagnosi alle consulenze specialistiche (Vol. 1). Piccin.
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *