01 dicembre 2024
Alcune mutazioni dei geni globinici (sostituzioni, micro-delezioni, inserzioni) possono produrre difetti strutturali caratterizzati dalla sintesi di catene globiniche che non consentono la formazione di tetrameri stabili. Tali difetti sono descritti associati a gradi diversi di instabilità e costituiscono il gruppo delle così dette «varianti instabili dell’emoglobina».
Questi particolari difetti dell’emoglobina (Hb) sono all’origine di processi di denaturazione e precipitazione presentando una ridotta sopravvivenza eritrocitaria. Di conseguenza si potranno avere anemie emolitiche croniche o acute in varie circostanze e forme. Questi fenomeni si osservano normalmente "in vivo" o anche "in vitro" mediante test dedicati. Le Hb instabili esprimono un fenotipo clinico che si trasmette con un meccanismo autosomico dominante che, in alcuni casi come accade per le varianti instabili delle catene beta globiniche, viene assimilato alle talassemie intermedie, particolarmente quando queste varianti Hb vengono co-ereditate con difetti talassemici a formare composti eterozigoti. Le varianti instabili delle catene β ad oggi osservate sono oltre 100, sono difetti rari documentati nella maggior parte dei casi in singoli soggetti (tra cui molti casi de novo), oppure in nuclei familiari isolati. Tuttavia, alcune di queste varianti instabili come l’Hb Köln, l’Hb Genova o l’Hb Zurigo risultano essere più frequenti e presenti in popolazioni diverse.
Le varianti instabili che interessano i geni a (poco più di 40) verranno considerate successivamente. Non si parla di solito di varianti instabili associate ai geni δ per l’assenza di espressioni fenotipiche caratteristiche dovuta alla bassa capacità sintetica del gene δ; in pratica si può solo classificare instabile un difetto δ partendo dal tipo di sostituzione amminoacidica coinvolta o dalla bassa percentuale di Hb A2-X prodotta. Rari sono i difetti dei geni γ con caratteristiche instabili riscontrate in neonati con ittero. Si può quindi osservare che la maggior parte delle varianti instabili presentano un difetto sulle catene β.
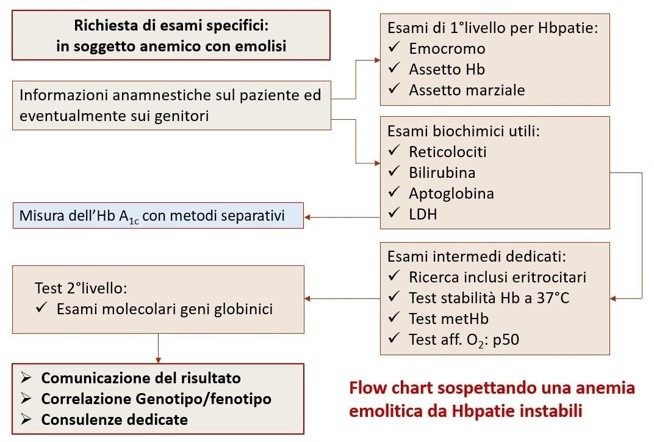
In generale, l’individuazione delle varianti Hb instabili impegna il laboratorio con esami mirati, utili a discriminare tra le possibili diverse cause di emolisi (1-3), esami ancor oggi non sostituibili anche dopo una caratterizzazione molecolare.
Questo «caso 15» vuole porre l’attenzione su tre pazienti che presentano tre varianti instabili delle catene β con caratteristiche diverse. Le tre situazioni qui esposte sono state gestite sulla traccia della flow chart qui riportata utilizzando anche ‘homemade methods’ ancora oggi non sostituibili e ancora consigliati per dare una veloce ed efficace risposta alle richieste del clinico che può aver osservato e segnalato utili indizi. Nei pazienti qui riportati si è giunti all’analisi molecolare omettendo alcuni esami funzionali ma che potrebbero essere eseguiti successivamente per una migliore gestione della consulenza genetica alla luce del difetto riscontrato.

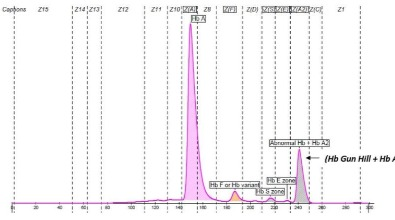
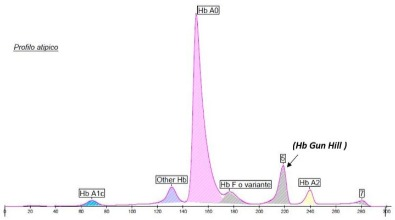
L’Hb Gun Hill è una variante delle catene β associata a lieve emolisi compensata. La catena anomala βGun Hill presenta una delezione di cinque aminoacidi tra i codoni 91 e 95. Ciò comporta una instabilità che si accompagna alla tendenza a formare dimeri che si separano a pH diversi. La delezione comprende l’His prossimale che lega l'eme in posizione 92 che porta ad una affinità alterata per l’O2 (4).
HBB:c.274_288delCTGCACTGTGACAAG;
β 91(F7) - 95(FG2) Leu-His-Cys-Asp-Lys->0
(Hb Gun Hill) eterozigote
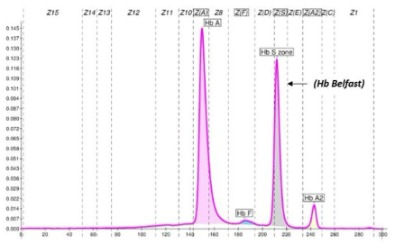
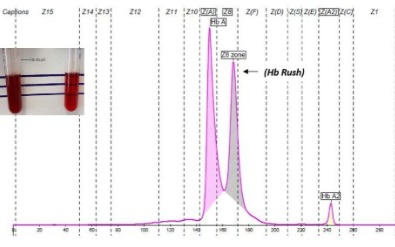
L’Hb Belfast è una variante instabile che si denatura sotto l’azione di sostanze o farmaci ossidanti con crisi emolitiche acute.
E’ stata descritta in famiglie di etnie diverse (5).
HBB: c. 46T>A; β 15(A12) Trp>Arg
(Hb Belfast) eterozigote
L’Hb Belfast è una variante instabile che si denatura sotto l’azione di sostanze o farmaci ossidanti con crisi emolitiche acute.
E’ stata descritta in famiglie di etnie diverse (5).
HBB: c. 46T>A; β 15(A12) Trp>Arg
(Hb Belfast) eterozigote
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *