01 febbraio 2024
INTRODUZIONE
Nel «Caso 5» di questo portale, si è visto come poter escludere la presenza di β talassemia in tre soggetti portatori di varianti delle catene β globiniche.
Nel «Caso 11», con riferimento ai valori ridotti dell’Hb A2, si è considerato l’effetto prodotto dalla presenza di varianti delle catene a e δ sul valore dell’Hb A2, quando si vuole escludere o ipotizzare la presenza di un tratto β talassemico.
Con altri esempi, si torna qui sul significato da attribuire all’Hb A2, all’Hb F e all’MCV ridotto in relazione all’eventuale co-ereditarietà di difetti β talassemici quando sono presenti varianti delle catene β globiniche. In particolare, a tale proposito ci chiediamo:
INTERPRETAZIONI
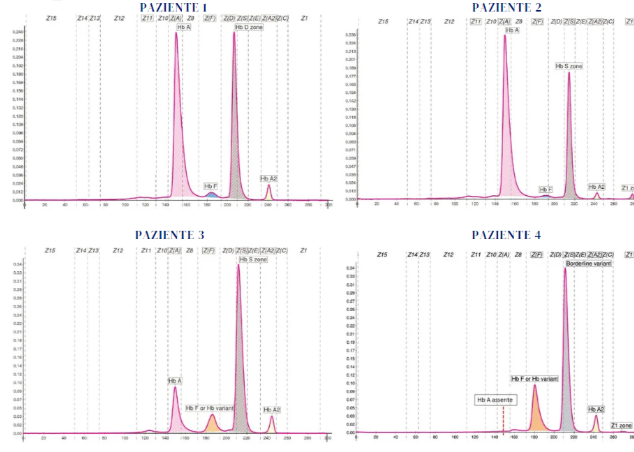
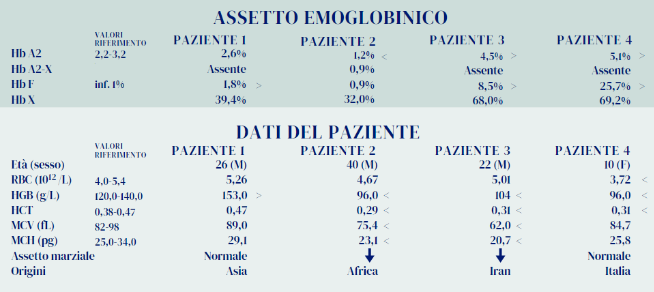
Paziente 1)
HBB: c.364G>C; β 121(G6) Glu>Gln (Hb D Punjab) eterozigote
HBG2:c.-211 C>T; -158 C>T eterozigote
Paziente non trasfuso. Il profilo elettroforetico mostra un picco che può essere facilmente attribuito ad una variante β soprattutto per la percentuale relativa e l’assenza di uno sdoppiamento dell’Hb A2. I parametri eritrocitari fanno escludere la presenza di difetti talassemici. L’Hb F, appena sopra i valori di riferimento, trova spiegazione nella presenza del polimorfismo associato al gene HBG2 rilevato durante l’analisi molecolare eseguita per la caratterizzazione della variante Hb.
Paziente 2)
HBB: c.20A>T; β 6(A3) Glu>Val (Hb S) eterozigote
HBD: c.49 G>C; δ 16(A13) Gly>Arg (Hb A’) eterozigote
Paziente non trasfuso. Presenza di Hb S confermata con test di sickling dal 1°livello. Hb A2: avendo definito la presenza di una variante beta, l’evidente sdoppiamento dell’Hb A2 deve essere attribuito ad una mutazione dei geni δ. L’Hb S risulta in percentuale più bassa rispetto ai valori medi riscontrati per le varianti β, ciò può essere attribuito all’anemia sideropenica. L’anemia incide anche sulle quantità relative dell’Hb A2 e dell’Hb A2-X. La caratterizzazione molecolare dei difetti riscontrati conferma i dati del 1°livello ed evidenzia la presenza della variante δ più conosciuta e diffusa in Africa.
Paziente 3)
HBB: c.20A>T; β 6(A3) Glu>Val (Hb S) eterozigote
HBB: c.92+6 T>C; IVSI-6 (β+ tal) eterozigote
Paziente non trasfuso. Presenza di Hb S confermata con test di sickling dal 1°livello. L’Hb A risulta pari al 19,0%, pertanto si deve ipotizzare la presenza di un difetto beta+ associato all’Hb S. Il quadro di una talassodrepanocitosi (composto eterozigote per Hb S/ Beta talassemia) è coerente con l’alterazione dei parametri eritrocitari e dell’assetto Hb, compresa l’Hb F (vedere dati in Tabella). La caratterizzazione molecolare è necessaria per definire o confermare il fenotipo ipotizzato.
Paziente 4)
HBB: c.20A>T; β 6(A3) Glu>Val (Hb S) eterozigote
HBB: c.118 C>T; codon 39 (β0 tal) eterozigote
Paziente non trasfuso. Presenza di Hb S confermata con test di sickling dal 1°livello. L’Hb A è assente, ciò può far ipotizzare la presenza di Hb S omozigote o di un difetto β0 associato all’Hb S. L’Hb A2 sensibilmente aumentata fa propendere per un composto eterozigote per Hb S/ β0 talassemia. La caratterizzazione molecolare è necessaria per definire o confermare il fenotipo ipotizzato.
CONSIDERAZIONI CONCLUSIVE
Una trasfusione, infatti, comporta la presenza di «sangue normale» che altera i rapporti percentuali relativi tra le diverse Hb, fisiologiche e/o patologiche presenti nel soggetto in esame.
a) conferma con test specifico (test di falcizzazione per Hb S)
b) conferma con altro metodo separativo (solo per Hb C e Hb E)
c) l’Hb A2 non è sdoppiata
d) la variante β è compresa fra 30 e 48% in assenza di microcitosi (eccetto il caso di varianti instabili)
Consideriamo l’esempio di un soggetto eterozigote per Hb S, confermato con test di falcizzazione: uno dei due geni β è mutato, con un difetto al codone 6 Glu>Val (Hb S) e consentirà quindi la sintesi di catene β-S mentre l’altro gene β, sull’altro cromosoma, è normale e produrrà catene β-A. Il risultato è quello di avere la sintesi di Hb A e di Hb S in un rapporto teorico relativo di 1:1. In realtà la sintesi delle due Hb è spostata a favore dell’Hb A per una maggior affinità delle catene β-A con le catene a rispetto alle β-S, quindi 55-65% di Hb A contro 32-45% di Hb S. Tale rapporto sbilanciato potrà variare secondo le caratteristiche della variante β presente. L’Hb A2, in soggetti eterozigoti per una variante β, nella maggior parte dei casi rimane entro i limiti della norma (inf. 3,2%). Raramente si possono osservare lievi incrementi dell’Hb A2. Tuttavia, in presenza di una variante β, tali incrementi non provano l’esistenza di difetti β talassemici se la percentuale dell’Hb A è superiore al 50%.
a) valori di Hb A2 inferiori a 1,5-1,7% possono indicare l’assenza di difetti β talassemici in soggetti con varianti (a o β) che si separano in Z15 o Z14.
b) un valore di Hb A2 superiore a 2,0% in un soggetto con microcitosi può essere osservato con una variante a associata a β talassemia.
c) un valore di Hb A2 compreso fra 2,5 e 3,5% può indicare, in assenza di microcitosi, una variante β non associata a β talassemia.
BIBLIOGRAFIA
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *