01 febbraio 2025
Nel precedente «caso 15» sono state considerate le varianti instabili delle catene β e la loro rilevanza nel contesto delle anemie emolitiche. Questo «caso 16» si sofferma a valutare in modo distinto le varianti instabili delle catene α cercando di interpretare alcune espressioni fenotipiche meno marcate che tuttavia il laboratorio può rilevare. Occorre premettere che anche nel caso delle varianti α instabili siamo di fronte a variazioni che interessano particolari sostituzioni amminoacidiche e particolari siti critici delle catene globiniche. Si deve ricordare che le catene α globiniche sono prodotte in quantità diversa dai geni HBA2 e HBA1 su ciascun cromosoma 16. Ciò rappresenta una delle cause dell’eterogeneità dei fenotipi osservabili che possono apparire anche «silenti» o comunque meno rilevanti soprattutto allo stato eterozigote rispetto alle forme classiche prodotte dalle varianti instabili delle catene β.Nel caso delle varianti α instabili i fenomeni emolitici caratteristici sono rilevabili prevalentemente "in vitro" mediante test dedicati, pertanto nella maggior parte dei casi, in assenza di altri segni, si arriva a determinare l’instabilità di una variante solo seguendo un percorso diagnostico standardizzato, come quello riportato nel «caso 15». Pertanto, anche in mancanza di evidenti condizioni emolitiche, quando viene osservata una nuova variante, è buona regola procedere alla caratterizzazione della stabilità strutturale mediante i tradizionali test «in vitro». Sono meno di 40 le varianti α instabili che ad oggi risultano riportate nei database dedicati (1) ma tale numero deve essere considerato sottostimato a causa della mancata specifica caratterizzazione della stabilità strutturale di molte nuove varianti osservate soprattutto nel corso degli anni recenti. Questo «caso 16» pone l’attenzione su due donne in gravidanza. Il laboratorio è stato chiamato in causa per accertare l’eventuale presenza di difetti talassemici nelle due Pazienti nelle quali era stata diagnosticata in precedenza una anemia microcitica con ferro normale. I relativi parametri eritrocitari ed emoglobinici sono riportati in Tabella.

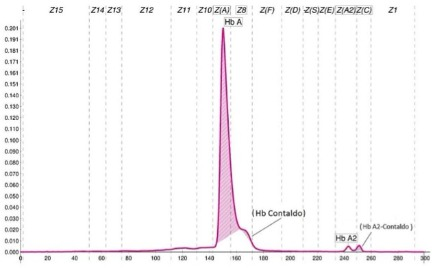
La variante Hb Contaldo (2) allo stato eterozigote è descritta come moderatamente instabile (1).In questa Paziente la variante è presente in associazione all’α-talassemia, ciò contribuisce ad esaltarne il comportamento instabile e la ridotta sopravvivenza eritrocitaria. La madre della Paziente, che non è portatrice di α-talassemia, non presenta segni di emolisi.
•La variante non si separa nettamente da Hb A e si presenta in quantità inferiore al 10%, tale quantità è significativamente bassa nonostante la presenza dell’α-talassemia.
•L’Hb A2-Contaldo appare invece in quantità relativa simile all’HbA2-Normale, segno di una maggior stabilità del tetramero α2Contaldoδ2 rispetto al tetramero normale α2δ2 .
•Nella Paziente, che è in gravidanza, i due difetti sono presenti su alleli diversi e non verranno quindi trasmessi assieme.
•Il neonato avrà 50% di probabilità di ereditare la variante HbContaldo che si presenterà in una forma moderatamente instabile (il partner della Paziente non è portatore di difetti globinici). Alla nascita tuttavia è possibile che il tetramero α2Contaldoγ2 possa risultare più instabile e produrre una condizione itterica nel neonato fino al completamento dello switch dell’Hb F dopo i sei mesi d’età.
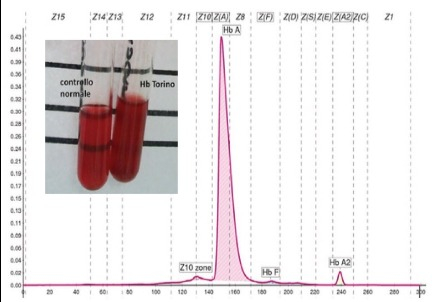
•La variante Hb Torino (3) è una α-variante descritta la prima volta in una famiglia di Treviso migrata in Piemonte ed in seguito osservata in alcune famiglie del Veneto apparentemente non imparentate tra loro. Un caso isolato ritenuto de novo è stato descritto in un soggetto libanese. I vari casi studiati hanno dimostrato fenotipi clinici diversi non chiariti nel passato.
•L’osservazione di comportamenti emolitici più marcati ha trovato spiegazione recentemente nella co-eredità dell’Hb Torino con l’ α talassemia.
•È il caso di questa Paziente 2 che in età adolescenziale era stata anche splenectomizzata nella speranza di poter migliorarne il quadro clinico.
•La variante non si separa né in CE né in HPLC.
•La presenza della variante instabile è stata documentata mediante il test di instabilità all’isopropanolo (vd. Figura con controllo normale).
•Il profilo elettroforetico ottenuto per la misura dell’Hb A1c ne ha evidenziato un valore significativamente basso (12 mmoli/moli), dovuto alla ridotta sopravvivenza eritrocitaria dell’Hb Torino.
1.Giardine B, Borg J, Viennas E, Pavlidis C, Moradkhani K, Joly P, et al. Updates of the HbVar database of human hemoglobin variants andthalassemia mutations. Nucleic Acids Res. 2014 Jan;42 (Database issue): D1063-9. http://globin.cse.psu.edu/hbvar/menu.html. (Accesso: Aprile2023).
2.Sciarratta GV, Ivaldi G, Molaro GL, Sansone G, Salkie ML, Wilson JB, Reese AL, Huisman TH. The characterization of hemoglobin Manitoba or alpha(2)102(G9)Ser>Arg beta 2 and hemoglobin Contaldo or alpha (2)103(G10)His>Arg beta 2 by high performance liquid chromatography.Hemoglobin. 1984;8(2):169-81.
3.Prato V, Gallo E, Ricco G, Mazza U, Bianco G, Lehmann H. Haemolytic anaemia due to haemoglobin Torino. Br J Haematol. 1970 Jul;19(1):105-15.
4.Barberio G, Ivaldi G. Varianti instabili dell’emoglobina: una sfida per il laboratorio? Biochimica clinica; 2021. DOI:10.19186/BC_2022.001.
5.Barberio G, Ivaldi G. (2020). Emoglobinopatie. Dalla diagnosi alle consulenze specialistiche . Piccin.
6.www.site-italia.org: Diagnostica di I e II livello delle Emoglobinopatie Buone Pratiche SITE, 2022.
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *