01 novembre 2022
INTERPRETAZIONI
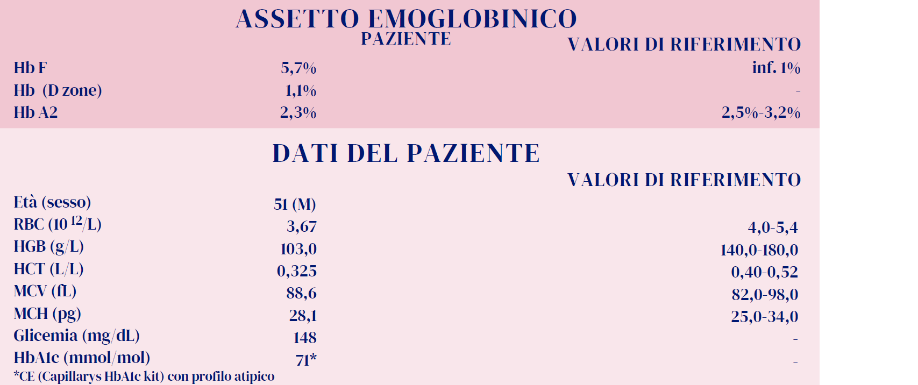
Il paziente perviene al laboratorio con richiesta di esami per glicemia, Hb A1c ed emocromo.
IPOTESI
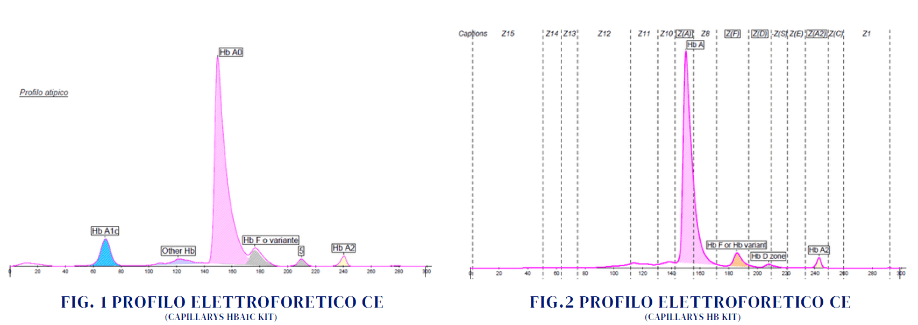
Se osserviamo la Fig.1 e la Fig.2 si può ipotizzare la presenza di un qualche polimorfismo che determina la sintesi persistente di Hb F.
Al picco minore in «D zone» potremmo attribuire almeno 3 significati diversi:
APPROFONDIMENTI
Le ipotesi fatte hanno trovato un riscontro nell’esame molecolare che ha evidenziato la presenza del composto eterozigote Beta talassemia/Hb Lepore (HBB:c.118C>T/NG_000007.3:g.63632_71046del) nel paziente che è risultato quindi trasfuso. Un soggetto adulto, non trasfuso, con questi difetti è visibile in Fig. 3 e lo schema di Fig. 4 illustra tale condizione alla nascita. Il difetto β0 tal (cod 39) riscontrato non consente, assieme all’Hb Lepore, la sintesi di Hb A. Pertanto la misura dell’Hb A1c effettuata in prima battuta nel paziente è stata fatta sull’Hb A del sangue trasfuso, fornendo un dato ingannevole. In mancanza di ogni informazione, aver posto un problema sulla presenza di frazioni Hb aggiuntive è stato fondamentale. Con un metodo non separativo questi allarmi non sarebbero stati disponibili.
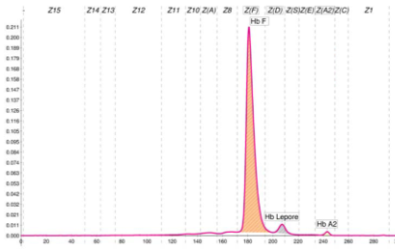
Fig. 3 Elettroferogramma di un individuo adulto non trasfuso con Hb Lepore/β0-Talassemia
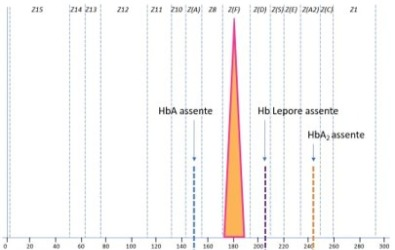
Fig. 4 Schema di elettroferogramma riscontrabile alla nascita in presenza di Hb Lepore/β0 -Talassemia
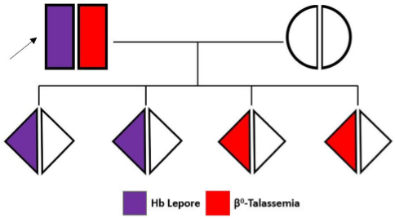
Questo composto Beta talassemia/Hb Lepore è statoriportato in letteratura associato a fenotipi variabilmentemarcati e non sempre trasfusione dipendenti, ciòsoprattutto in relazione al tipo di beta talassemia presente.Un soggetto come quello qui descritto, con partner normale,avrà la possibilità di vedere i suoi difetti segregati in tutti glieventuali figli.Alla nascita, la presenza di questi difetti produrranno unfenotipo sovrapponibile a quello del Morbo di Cooley.
CONCLUSIONI
BIBLIOGRAFIA
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *