01 settembre 2022
INTERPRETAZIONI
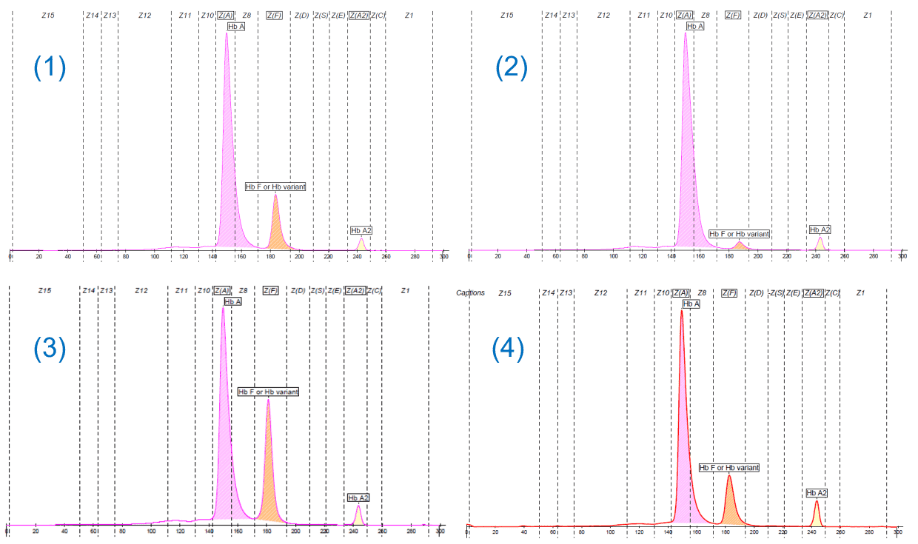
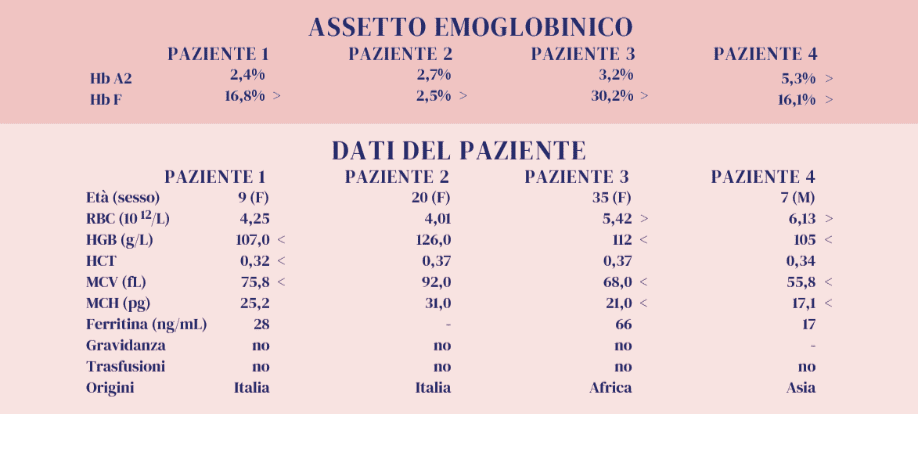
Obiettivo di questo «Caso Clinico» è quello di presentare quattro pazienti con fenotipi diversi caratterizzati dalla presenza di Hb F variabilmente espressa, in soggetti adulti. L’interpretazione dei livelli di Hb F, sulla base degli esami di 1° livello, può suggerire ipotesi diagnostiche che potranno richiedere conferme molecolari.
Sappiamo che l’Hb F è costituita da due catene a e due catene γ. Queste ultime sono prodotte da due geni (HBG1 e HBG2) che funzionano tipicamente nell’epoca fetale mentre dopo la nascita si ha il loro rapido silenziamento a favore del gene β (HBB) con la produzione di emoglobina adulta (HbA). Nella norma i valori di Hb F sono inferiori all’ 1% (1). Tuttavia l’attività dei geni γ può persistere nella vita adulta con presenze piu’ o meno importanti di Hb F, segno distintivo di uno spettro molto eterogeneo di condizioni (2-4):
Paziente 1. Microcitosi, Hb A2 nella norma e Hb F superiore al 15% :
Data l’età potrebbe essere utile esaminare i genitori, l’esame molecolare è comunque necessario.
Paziente 2. Modesto aumento dell’Hb F in un quadro di normalità degli indici eritrocitari.
Paziente 4. Tutti i dati disponibili definiscono un quadro β-talassemico con una quota significativamente elevata di HbF. Si possono fare alcune ipotesi:
BIBLIOGRAFIA
1.Traeger-Synodinos, J., Harteveld, C.L., Old, et al. EMQN Best Practice Guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. Eur J Hum Genet 2015;23:426-37.
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *