01 luglio 2022
INTERPRETAZIONI
Obiettivo di questo «caso clinico» è quello di giungere a conclusioni diagnostiche mediante esami di 1° livello per le emoglobinopatie eseguiti alla nascita. Vengono riportati i casi di tre neonati, nati a termine ed esaminati in modo appropriato entro la prima settimana di vita, così come previsto dalle linee guida (1): in nessun caso, per i tre neonati, era stata eseguita la diagnosi prenatale.
Neonato 1
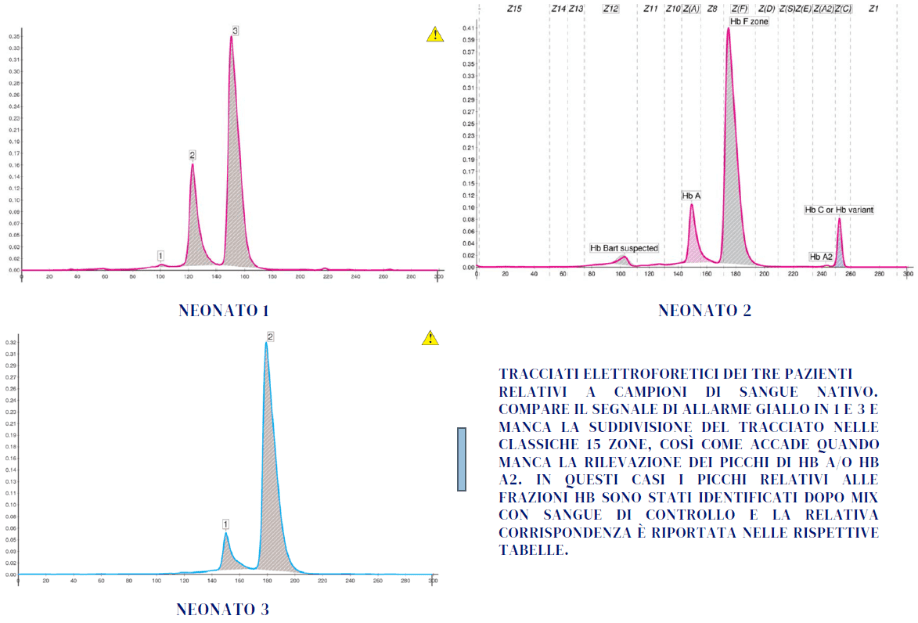
Genitori entrambi portatori eterozigoti di Hb S: l’esame alla nascita ha mostrato l’assenza di tale variante e lo stato di normalità per il neonato, con un valore di Hb A > 22% (1). È certa in questo caso l’assenza nel neonato dell’Hb S, sia allo stato eterozigote che omozigote.
Neonato 2
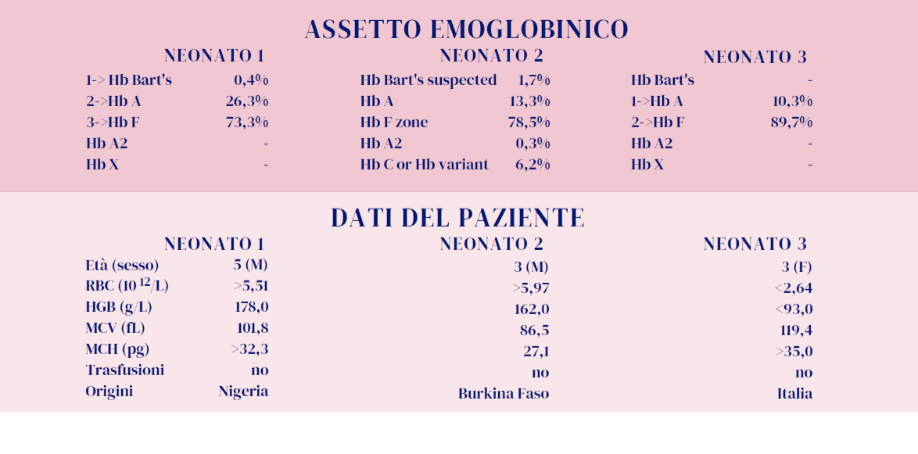
Genitori portatori rispettivamente di Hb C e β-talassemia: l’esame alla nascita del neonato indica chiaramente la presenza dell’Hb C e dell’Hb A, conseguentemente la sua condizione è quella di un portatore eterozigote di Hb C (quindi senza β-talassemia).
Probabilmente è presente un tratto a-talassemico, caratteristica non indagata nei genitori, che si evidenzia con la presenza di una quota di Hb Bart’s quantificata dall’esame elettroforetico. La presenza di a-talassemia non pregiudica il fenotipo clinico del neonato.
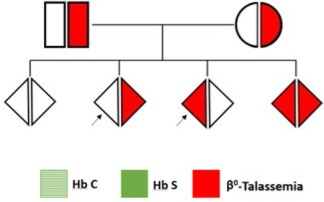
Neonato 3
Genitori entrambi portatori di β°-talassemia: l’esame alla nascita è stato eseguito con un duplice obiettivo: quello di tranquillizzare la coppia che non aveva fatto diagnosi prenatale e di valutare l’idoneità del sangue del cordone ombelicale, in vista del bancaggio, considerando l’intenzione espressa dai genitori di donarlo. Il neonato risulta anemico, ma la presenza dell’Hb A rassicura sullo stato di «non-omozigosi» della β-talassemia. Probabilmente è presente uno stato di talassemia eterozigote. Tale condizione, che non desta alcuna preoccupazione, deve essere confermata, così come previsto dalle linee guida (1,2), attendendo il completamento dello switch dell’Hb F o mediante specifico esame del DNA. Per quanto riguarda il bancaggio, il sangue cordonale può essere conservato in attesa della conferma diagnostica successiva (3).
CONCLUSIONI
Le raccomandazioni per la diagnosi neonatale delle emoglobinopatie (1) definiscono i limiti e le possibilità diagnostiche mediante esami di 1° livello, talvolta eseguiti alla nascita in particolari condizioni di urgenza.
In sintesi possiamo osservare che:
BIBLIOGRAFIA
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *