01 dicembre 2021
INTERPRETAZIONI
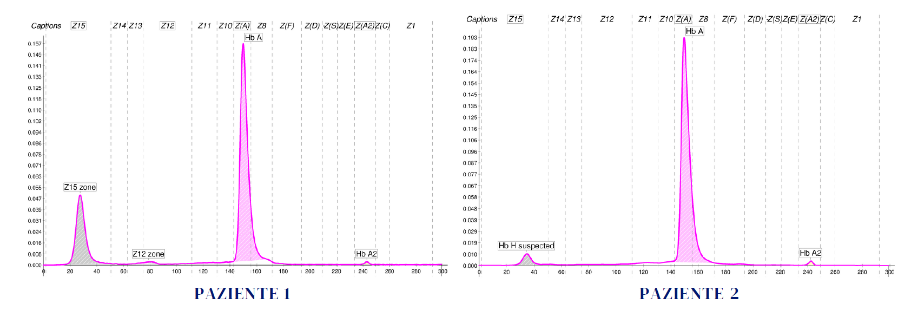
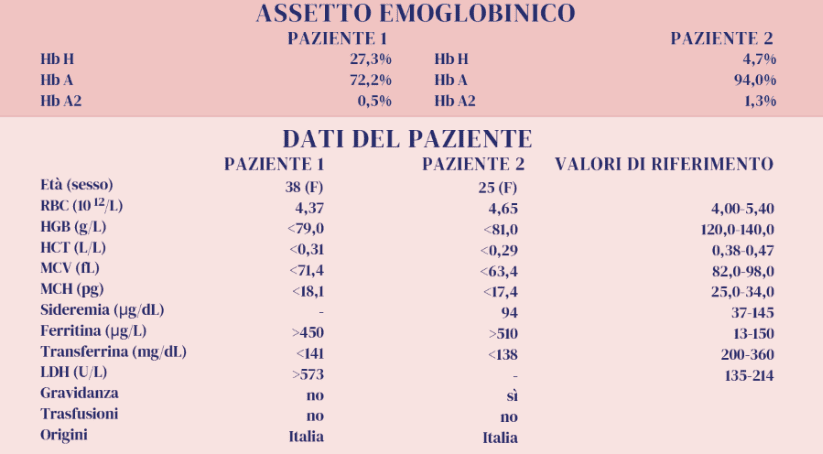
Vengono illustrati due quadri emoglobinici relativi a due giovani donne (Paziente 1 e Paziente 2). Gli assetti emoglobinici mostrano per entrambe la presenza di un picco anomalo in Z15, zona nella quale si sospetta in primis la presenza di Hb H. Ciò che appare evidente da subito è il diverso valore di tali frazioni emoglobiniche e il basso valore delle Hb A2.
IPOTESI
I dati disponibili e i risultati degli esami eseguiti, pur consentendo al laboratorio di poter concludere indicando la presenza di Hb H in entrambe le pazienti con pochi margini di incertezza, ci danno la possibilità di riflettere sulle ipotesi a favore di tali conclusioni e sui perché di alcune differenze. L’Hb H si evidenzia normalmente con i diversi metodi separativi in quantità variabile, in relazione ai difetti talassemici presenti e alla denaturazione dell’Hb H.
CONCLUSIONI
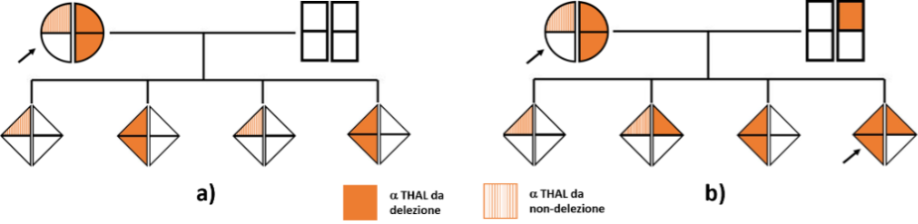
Gli esami eseguiti e le poche informazioni anamnestiche disponibili hanno consentito di formulare ipotesi che poi si sono rivelate corrette. Da notare che, nel caso della paziente 2, lo stato di gravidanza ha accentuato la condizione di anemia rendendola più marcata di quella normalmente attesa. Viene ulteriormente dimostrata l’utilità di una consulenza genetica supportata da una caratterizzazione molecolare dei difetti coinvolti (5).
BIBLIOGRAFIA
1.Wajcman H, Traeger-Synodinos J, Papassotiriou I, et al. Unstable and thalassemic alpha chain hemoglobinvariants: a cause of Hb H disease and thalassemia intermedia. Hemoglobin 2008;32:327-49.
2.Higgs DR, Weatherall DJ. The alpha thalassaemias. Cell Mol Life Sci 2009; 66: 1154–62.
3.Harteveld and Higgs, Alpha-thalassaemia Orphanet Journal of Rare Diseases. 2010, 5:13
4.HbVar database for human hemoglobin variants and thalassemia mutations.http://globin.bx.psu.edu/hbvar/menu.html bx.psu.edu (ultimo accesso 29.10.2021).
5.Barberio G, & Ivaldi G. (2020). Emoglobinopatie. Dalla diagnosi alle consulenze specialistiche (Vol. 1). Piccin
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *