01 novembre 2021

INTERPRETAZIONI
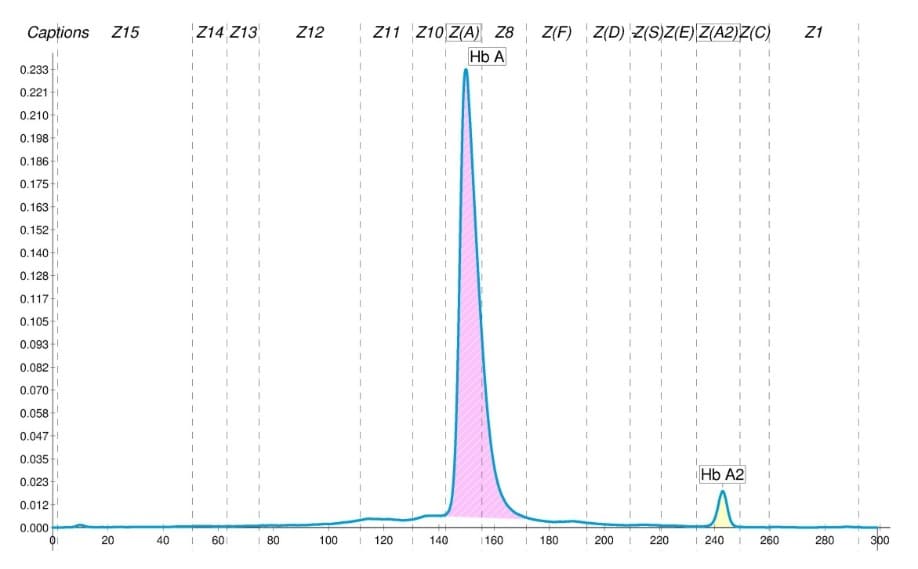
Si tratta di una ragazza alla quale sono stati prescritti esami per la ricerca di eventuali emoglobinopatie. Gli esami definiscono le caratteristiche quali-quantitative dell’assetto emoglobinico (Hb), i parametri eritrocitari e l’assetto marziale, così come previsto dalle linee guida per il 1° livello diagnostico (1,2). Alcuni parametri appaiono "moderatamente" fuori norma. Le informazioni anamnestiche disponibili non sono complete. Alcune ipotesi potranno essere formulate.
IPOTESI
L’emoglobina A2 (HbA2), nell’ambito degli esami di primo livello, rappresenta il più importante parametro diagnostico utile per individuare il portatore di β-talassemia. L’Hb A2può essere considerata normale quando presenta valori compresi tra 2,5 e 3,2 % (1).Tuttavia, valori più bassi (da 2 a 2,5%) o più alti (da 3,3 a 3,7%), borderline rispetto ai valori di riferimento, possono essere associati a varie condizioni, anche normali, da valutare sempre attentamente. Una misura dell’Hb A2 superiore al 4,5% è riscontrabile prevalentemente in soggetti portatori di β0-talassemia, prodotta da difetti globinici cosiddetti "classici".
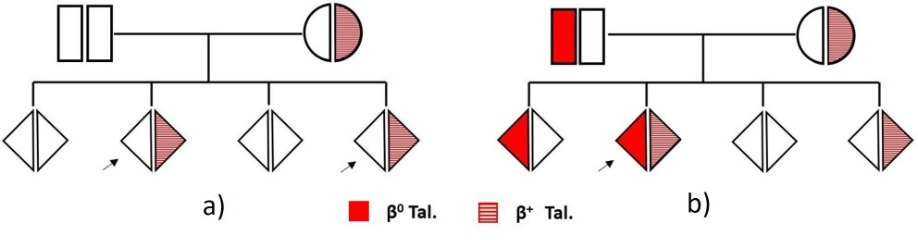
Tali valori contribuiscono alla diagnosi della condizione β-talassemica senza dover ricorrere alla conferma molecolare (1-3).Il caso qui rappresentato ci consente di fare le seguenti considerazioni:• È molto probabile che il valore di Hb A2 (3,9%) e la microcitosi riscontrati in questo caso, siano il risultato di un difetto talassemico più lieve definito β+-talassemia. La paziente, con un eventuale futuro partner non portatore di difetti globinici (Fig. a), avrebbe il 50% di probabilità di trasmettere ai figli il tratto talassemico. Se invece il partner dovesse essere portatore di β0-talassemia vi sarebbe la probabilità che il 25% dei figli ricevano entrambi i difetti dei genitori (Fig. b). In tal caso la condizione clinica più importante che si potrebbe osservare sarebbe quella di una "talassemia intermedia" caratterizzata da anemia e segniclinici variabilmente marcati, di solito non trasfusione dipendenti (4-6).Altre ipotesi meno probabili:• Considerando sempre l’Hb A2, l’MCV e l’MCH, potremmo anche ipotizzare la presenza di undoppio difetto δ+β-talassemico ovvero una δ+-talassemia associata ad una β0-talassemia.Questa combinazione è rara e si ha quando un difetto talassemico β0 "classico" può esprimere una Hb A2 ridotta per la presenza della δ+-talassemia (diminuita sintesi di cateneδ) (3,4). Questa possibilità presenta aspetti importanti dal punto di vista dell’interpretazione dei risultati e quindi della valutazione nell’ambito di una consulenza genetica.• Una situazione ancor più rara è quella che si può ipotizzare con la presenza di un difetto globinico instabile prodotto da varianti β che possono presentarsi elettroforeticamente silenti. Queste varianti instabili si associano talvolta ad un modesto incremento dell’Hb A2(7), mentre la microcitosi potrebbe, in questo caso, essere prodotta da a-talassemia coereditata (quest’ultima caratteristica è molto frequente nelle popolazioni Mediterranee, Africane ed Asiatiche).
CONCLUSIONI
Considerando tutti gli esami eseguiti e le informazioni anamnestiche disponibili si può ritenere molto probabile, nel caso trattato, la presenza di una β+-talassemia che richiederà comunque una opportuna conferma molecolare. Considerando l’età e le condizioni della paziente l’esame del DNA può anche essere differito nel tempo o con l’urgenza valutata dal medico curante secondo le circostanze.
BIBLIOGRAFIA
1.Raccomandazioni per la diagnostica di primo livello delle emoglobinopatie-SITE 2012 http://www.siteitalia.org/collana_scientifica.php. (ultimo accesso aprile 2019).
2.Angastiniotis M, Eleftheriou A, Galanello R, et al. In: Old J, ed. Prevention of Thalassaemias and Other Haemoglobin Disorders: Vol. 1: Principles. 2nd ed. Nicosia, Cyprus; 2013.
3.Mosca A, Paleari R, Ivaldi G, et al. The role of Haemoglobin A2 testing in the diagnosis of thalassaemias and related haemoglobinopathies. J ClinPathol 2009; 62:13–7.
4.Giambona A, Passarello C, Vinciguerra M, et al. Significance of Borderline Hemoglobin A2 Values in an Italian Population with a High Prevalence of beta-Thalassemia, Haematologica 2008; 93:1380-4.
5.Thein SL. The molecular basis of β-thalassemia. Cold Spring Harb Perspect Med 2013;3: a011700.
6.Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet 2018;391:155-67.
7.Ivaldi G, Barberio G, Harteveld C, et al. Hb A2 measurements in beta-thalassemia and in other conditions. Thalassemia Rep 2014; 4:45–8.
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *