03 aprile 2023
La presenza di un valore ridotto di Hb A2 può rappresentare un utile indicatore per segnalare la presenza di difetti globinici e non solo, per cui in molti casi richiede ulteriori indagini biochimiche o molecolari.
È importante inoltre ricordare che la corretta interpretazione della misura dell’Hb A2 e del suo possibile significato clinico non può mai prescindere dalla conoscenza di altri parametri analitici fondamentali forniti dall’emocromo e dall’assetto marziale, come indicato dalle suddette raccomandazioni.
I quadri di seguito riportati rappresentano le circostanze più frequentemente associate a valori ridotti di HbA2 in soggetti adulti non trasfusi.
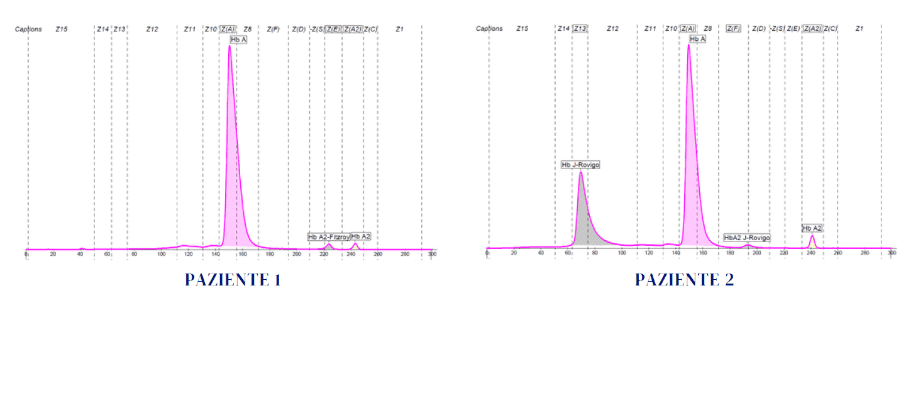
Hb A2 e presenza di varianti delle catene δ o di δ-talassemie. Quando uno dei due geni δ non consente la sintesi di catene utili alla formazione di Hb A2 (delta talassemia) o le produce mutate (varianti delta, α2 δ2X), si avrà un valore di Hb A2 ridotto fino al 50%, rispetto alla norma. Le varianti delta possono essere quantificate dai sistemi separativi utilizzati, oppure potrebbero essere non quantificabili perché co-migranti con Hb A o altre varianti eventualmente presenti. Talvolta l’Hb A2 potrebbe essere invece sintetizzata in misura tanto ridotta da risultare in quantità inferiore alla sensibilità della strumentazione utilizzata. Nel caso di δ talassemia, si osserva sempre e solo una riduzione dell’Hb A2 pari a circa il 50% rispetto al valore di riferimento normale. Nei rari casi in cui i geni δ presentano difetti in omozigosi (δ-talassemia omozigote), l’Hb A2 risulterà assente. Di seguito viene riportato un esempio in eterozigosi con alcuni parametri in tabella (paziente 1) ed il profilo elettroforetico (figura 1) che mostra la separazione della variante Hb A2-Fitzroy.
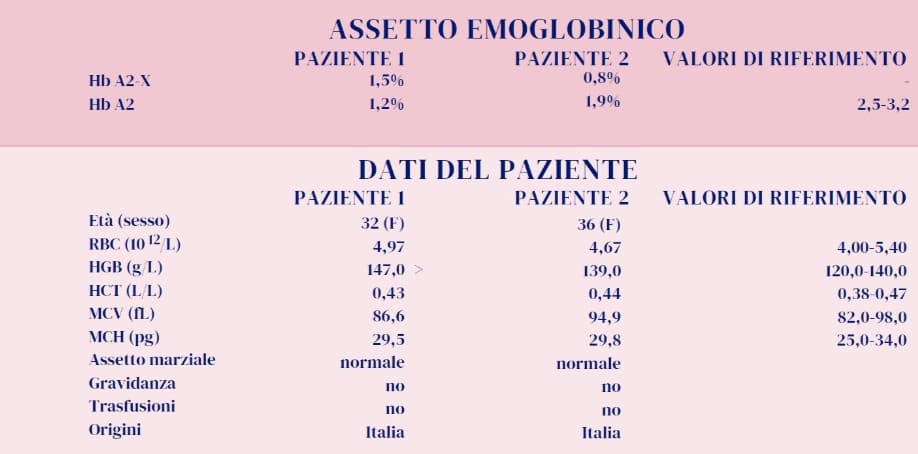
Hb A2 e varianti delle catene α. Altra circostanza nella quale possiamo riscontrare un valore sensibilmente ridotto dell’Hb A2 è la presenza di una variante delle catene α globiniche. Ciò è dovuto al fatto che anche le catene α contribuiscono a formare l’Hb A2 e quindi il tetramero globinico risentirà della presenza di tale mutazione (α2Xδ2). Vi sono circostanze in cui non è possibile osservare l’Hb A2 mutata (A2-X ); cio’ può accadere quando la variante α co-migra con l’Hb A o quando la variante è particolarmente instabile. Di converso occorre sottolineare che una eventuale contemporanea presenza di α talassemia, così come contribuisce ad incrementare la quantità relativa della variante α, incrementerà anche il valore relativo di Hb A2-X riducendo quello dell’Hb A2 normale. Viene qui riportato un esempio con alcuni parametri in tabella (paziente 2) ed il profilo elettroforetico (figura 2) da cui risulta la presenza dell’ Hb J Rovigo (variante α) e dell’Hb A2 Rovigo secondaria alla variante α.
CONCLUSIONI
I valori di Hb A2 inferiori a 2,5% devono essere valutati considerando le possibili cause di decremento sopra esposte. Tuttavia, altre cause più rare possono essere presenti. Per queste si rimanda alle raccomandazioni per la diagnostica di 1° livello (1).
È opportuno ribadire che l’intervallo di normalità dell’Hb A2 indicato dalle citate raccomandazioni rappresenta l’insieme dei valori normali riscontrabili nel 95% della popolazione. Tuttavia, così come si conoscono valori normalità «boderline alti», che occorre tener presente, confermare e caratterizzare nel corso degli esami di prevenzione della beta talassemia, esistono valori «borderline bassi» che possono rientrare nella normalità ma senza presentare significato clinico. La variabilità dell’Hb A2 nel formare il tetramero (α2 δ2) può anche essere in relazione alla competizione e alla conseguente diversa affinità reciproca di tutte le catene prodotte nel soggetto adulto (α, β e δ normali o mutate), così come accade per le varianti Hb in generale (3).
Nella pratica di laboratorio occorre considerare che:
BIBLIOGRAFIA
1.www.site-italia.org: Diagnostica di I e II livello delle Emoglobinopatie Buone Pratiche SITE, 2022.
2.Denic S, Agarwal MM, Al Dabbagh B, et al. Hemoglobin A2 Lowered by Iron Deficiency and α -Thalassemia: Should Screening Recommendation for β -Thalassemia Change? ISRN Hematology. 2013;2013:858294.
3.Mavilio F, et al. Post-translational control of human hemoglobin synthesis: the role of the differential affinity between globin chains in the control of mutated globin gene expression. Biochimica et Biophysica Acta. 1980 Dec;610(2):339-351.
4.Barberio G, Ivaldi G. (2020). Emoglobinopatie. Dalla diagnosi alle consulenze specialistiche . Piccin.
5.Ivaldi G, Barberio G, Harteveld C, et al. HbA2 measurements in beta-thalassemia and in other conditions. Thalassemia Rep 2014; 4:45–8.
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *