01 gennaio 2023
PREMESSA
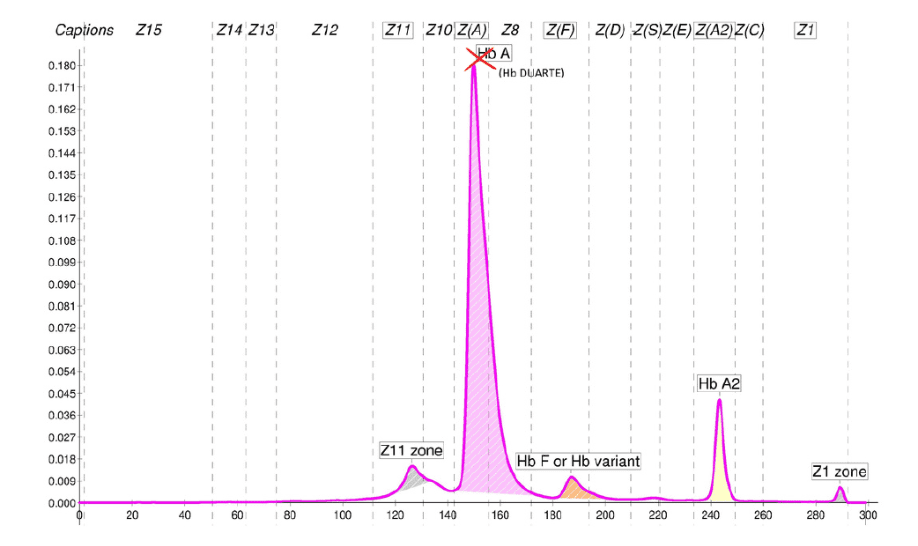
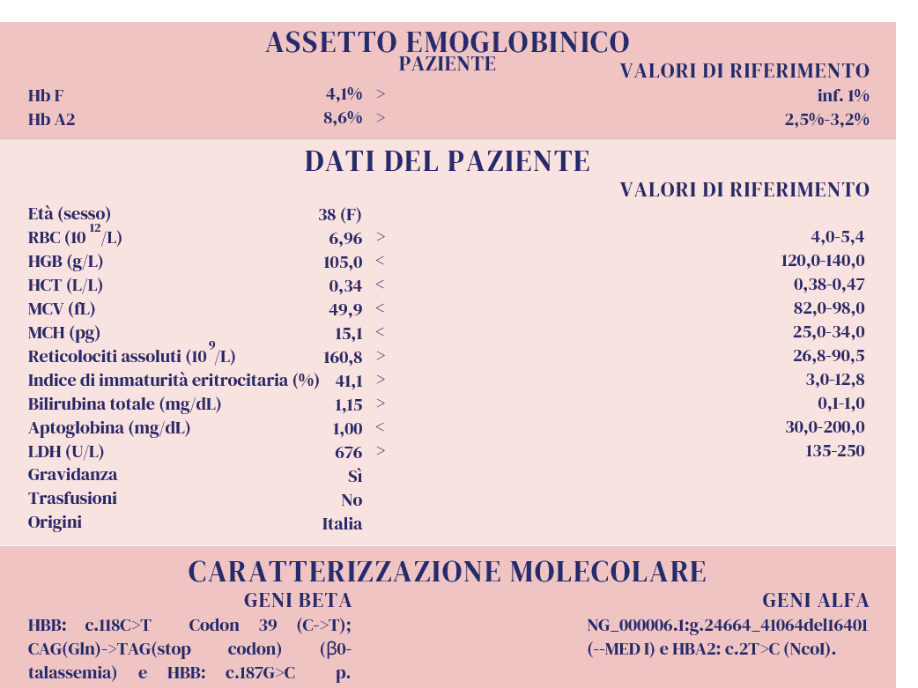
Il seguente caso ci consente di affrontare l’argomento dei valori superiori alla norma dell’Hb A2, in particolare di quelli più elevati. L’Hb A2 presenta valori considerati “nella norma”, dalle linee guida prodotte nel tempo, quando sono inferiori a 3,1-3,5 % (1,2), con una prevalenza maggiore di valori inferiori a 3,2%. Sappiamo che tali misure sono condizionate da numerosi fattori pre-analitici e analitici. L’imprecisione nella misura dell’Hb A2, anche valutata con sistemi separativi diversi, potrà essere ridotta quando materiali di controllo adeguati saranno definitivamente disponibili. Valori definiti “borderline” di Hb A2 cadono in un intervallo compreso tra 3,0 e 3,9 % e necessitano conferme, in genere molecolari, sulla presenza di difetti talassemici, soprattutto quando i parametri eritrocitari si presentano nella norma (3). Tuttavia, nella maggior parte dei casi, il valore aumentato dell’Hb A2 (tra 4,0% e 6,0%) consente di interpretare e definire correttamente almeno la presenza di una condizione eterozigote beta talassemica con variazioni riconducibili alla presenza di difetti β+ o β0 o di altri fattori modulanti (1). Talvolta, l’aumento della concentrazione relativa dell’Hb A2 si presenta talmente marcato ,“over the top”, da insinuarci dubbi, incertezza nell’interpretazione del dato e nelle conclusioni diagnostiche. Ciò ci riporta all’argomento di questo caso e alla ricerca delle possibili cause. Oltre alla presenza di un difetto β-talassemico vi sono altre rare condizioni che si accompagnano ad un aumento variabile dell’Hb A2 (4,5). Inoltre, dobbiamo sempre ricordare che la quantificazione dell’Hb A2, con qualsiasi metodo venga eseguita, fornisce sempre una «misura relativa». Facciamo ora l’esempio di ciò che si ha in un soggetto adulto con β0-talassemia eterozigote (β0-tal): abbiamo una capacità di sintesi dei geni β ridotta del 50% e una conseguente produzione di catene βA ridotte del 50% rispetto a quelle prodotte in un soggetto non portatore di β-talassemia, mentre le altre catene coinvolte nella sintesi dell’Hb A (α2 β2) e dell’Hb A2 (α2 δ2) vengono prodotte in quantità normale (in assenza di altri difetti). Considerando che il rapporto normale di sintesi tra le catene β e le catene δ è di circa 32:1, in presenza di β0-tal tale rapporto si dimezza passando a circa 16:1 (5) con conseguente variazione di espressione sia di Hb A che di Hb A2. L’Hb A2 passerà da un valore medio relativo pari al 3% del soggetto normale, ad un valore medio relativo aumentato a circa il 6 % del portatore di β0-talassemia. Valori più elevati di Hb A2, attorno al 7-9%, sono stati osservati in alcuni casi di β-talassemia dovuta ad elezioni del gene β globinico. Per tali valori elevati sono state fatte ipotesi con riferimento specifico alla rimozione di sequenze regolatrici o di regioni di controllo del gene δ. Si osserva però in letteratura che non tutti i soggetti che hanno tali delezioni presentano aumenti considerevoli dell’Hb A2 (6) quindi tale variabilità potrebbe anche essere riconducibile a co-fattori presenti e non indagati.
Il raro caso che qui viene riportato è stato studiato anche dal punto di vista molecolare e ci consente di fare alcune riflessioni basate sull’evidenza e formulare altre ipotesi sulle cause che possono rendere l’HbA2 particolarmente elevata.
IN SINTESI
IPOTESI & CONCLUSIONI
Nel caso qui documentato si osserva un valore insolitamente alto di Hb A2, perché?
BIBLIOGRAFIA
1.Mandrile G, Barella S, Giambona A, Gigante A, Grosso M, Perrotta S, Scianguetta S, Forni GL. First and Second Level Haemoglobinopathies Diagnosis: Best Practices of the Italian Society of Thalassemia and Haemoglobinopathies (SITE). J Clin Med. 2022 Sep 15;11(18):5426
2.Angastiniotis M, Eleftheriou A, Galanello R, et al. In: Old J, ed. Prevention of Thalassaemias and Other Haemoglobin Disorders: Vol. 1: Principles. 2nd ed. Nicosia, Cyprus; 2013
3.Giambona A, Passarello C, Vinciguerra M, et al. Significance of Borderline Hemoglobin A2 Values in an Italian Population with a High Prevalence of beta-Thalassemia, Haematologica 2008; 93:1380-4
4.Mosca A, Paleari R, Ivaldi G, et al. The role of Haemoglobin A2 testing in the diagnosis of thalassaemias andrelated haemoglobinopathies. J ClinPathol 2009; 62:13–7
5.Ivaldi G, Barberio G, Harteveld C, et al. Hb A2 measurements in beta-thalassemia and in other conditions. Thalassemia Rep 2014;4:45–8
6.Motum PI, Kearney A, Hamilton TJ, Trent RJ. Filipino beta zero thalassaemia: a high Hb A2 beta zero thalassaemia resulting from a large deletion of the 5' beta globin gene region. J Med Genet. 1993Mar;30(3):240-4
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *