01 ottobre 2021
.jpg)

INTERPRETAZIONI
Questo è uno dei casi, non rari, in cui la mancanza di informazioni anamnestiche non consente di trarre molte conclusioni dagli esami di 1° livello eseguiti. Tuttavia, se un test specifico per l’emoglobina S (test di sickling) venisse eseguito e risultasse positivo, potrebbe indicare con certezza la presenza di tale variante. Nel caso di negatività del test occorrerebbe eseguire necessariamente l’analisi molecolare per definire la variante e il caso nel suo insieme.
IPOTESI
L'assetto emoglobinico (Hb) e la percentuale relativa della variante suggeriscono di indagare se il paziente è stato sottoposto a trasfusione in tempi recenti. Inoltre, se si è accertata la presenza dell'Hb S, si possono fare alcune considerazioni:
- se negli ultimi tre mesi non sono state eseguite trasfusioni si può ipotizzare la presenza di un composto eterozigote Hb S/β-Talassemia, con un difetto talassemico di tipo β+ o β++(1). Tale ipotesi si basa sulla presenza dell’HbA e soprattutto sul dato percentuale della variante (Hb S) che risulta di poco, ma superiore al 50%. La caratterizzazione mediante analisi molecolare consente di definire con precisione la tipologia del difetto β-talassemico (β-tal.). Ciò è importante per la gestione clinica del paziente e per la definizione di una eventuale condizione «non-trasfusione-dipendente», quindi non assimilabile alla classica Sickle Cell Disease (SCD) (2,3).
- Se invece il paziente viene trasfuso regolarmente, o comunque lo è stato recentemente, si può pensare alla presenza di un composto eterozigote Hb S/β-Talassemia, con un difetto β-talassemico di tipo β di tipo β+ marcato (con una minima produzione di Hb A), oppure un classico difetto SCD con Hb S allo stato omozigote. Quest’ultima ipotesi è tuttavia molto meno probabile per la presenza di valori elevati di Hb A. L’analisi molecolare anche in questa circostanza sarà risolutiva nel percorso di caratterizzazione.
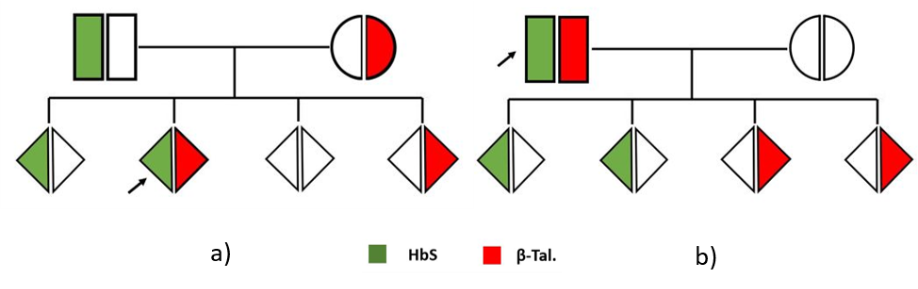
- Le risultanze del laboratorio richiedono il coinvolgimento del clinico che ha richiesto gli esami: è fondamentale infatti per il laboratorio reperire informazioni anamnestiche del paziente, ed eventualmente dei genitori, per la corretta interpretazione dei parametri riscontrati e l’adeguata conclusione del percorso analitico. Nel caso di genitori in cui siano noti i difetti qui ipotizzati (Hb S e β-tal), albero a), è indicato l’esame alla nascita dei figli che nel 25% dei casi potranno ricevere i difetti da entrambi i genitori.
Il composto eterozigote che abbiamo potuto descrivere in questo soggetto di 17 anni poteva già essere ipotizzato alla nascita (4). Non sappiamo se questo paziente sia stato effettivamente diagnosticato appena nato, ma soggetti con un fenotipo analogo o più marcato presentano caratteristiche che entro i primi mesi di vita, possono beneficiare di una profilassi adeguata mediante vaccinazione, atta a ridurre gli effetti prodotti da batteri capsulati che rappresentano un rischio per la vita dei neonati con queste patologie.
CONCLUSIONI
Osservando l’albero genealogico b), si comprende come il paziente potrebbe, con un partner non portatore di difetti emoglobinici, avere solo figli eterozigoti HbS o eterozigoti per β-talassemia. Tuttavia, è opportuno che la coppia, prima di una gravidanza, chiarisca tali concetti nell’ambito di una consulenza genetica specialistica.
BIBLIOGRAFIA
1.Divoky V, Baysal E, Schiliro G, et al. A mild type of Hb S /beta(+)-thalassemia [-92(C-->T)] in a Sicilian family. Am J Hematol 1993;42:225-6.
2.Russo G, De Franceschi L, Colombatti R, et al. Current challenges in the management of patients with sickle cell disease - A report of the Italian experience. Orphanet J Rare Dis. 2019;14:120.
3.AIEOP. Linee guida per la gestione della malattia drepanocitica in età pediatrica in Italia.2012
4.Ivaldi G, Barberio G, Caruso V, et al. Raccomandazioni per la diagnosi neonatale delle emoglobinopatie. Biochim Clin 2015; 39:116-34.INT
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *