01 maggio 2022

INTERPRETAZIONI
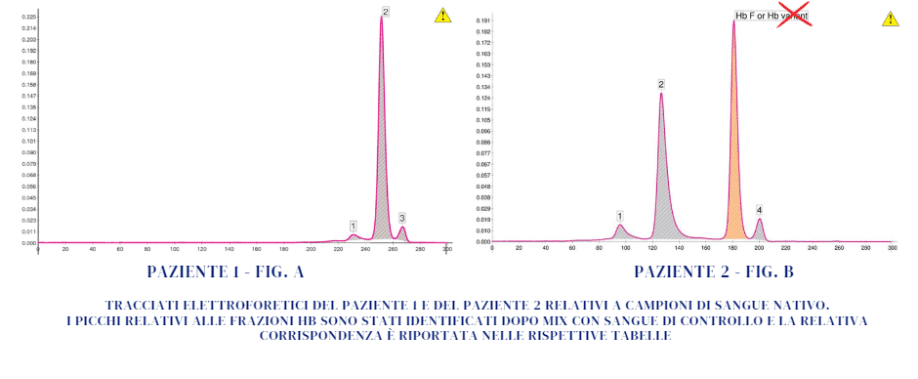
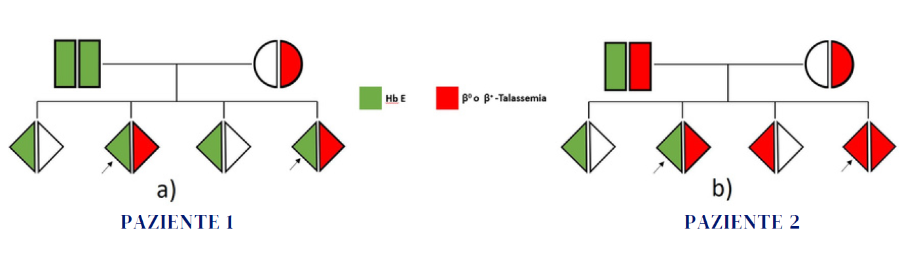
Con questo «caso clinico» si vuole evidenziare l’utilità degli esami di 1° livello nel riconoscere la condizione di omozigosi dell’Hb E da quella prodotta dal composto Hb E/ β-Talassemia. Vengono portati gli esempi di due pazienti, entrambi non trasfusi e originari del Bangladesh. I risultati degli esami riportati nelle due Tabelle sono stati ottenuti nel corso degli esami di 1°livello. Su tali risultati si possono basare alcune considerazioni certe, essendo nota la caratterizzazione molecolare di entrambi i pazienti. Paziente 1 è risultato portatore di Hb E allo stato omozigote [HBB:c.79G>A; β 26(B8) Glu>Lys]. L’Hb E è presente al 94%, associata a microcitosi e anemia assente (Fig. a). L’HbE è la seconda più conosciuta e diffusa variante emoglobinica dopo l’HbS ed è comune nel Sud-Est asiatico, dove la sua prevalenza può raggiungere il 30-40% in alcune parti della Thailandia, della Cambogia e del Laos, ma è molto presente anche nello Sri Lanka, India Nord-orientale, Bangladesh, Pakistan, Nepal, Vietnam, Malesia.
La catena mutata β-E viene sintetizzata ad una velocità ridotta rispetto a quella normale (β-A) poiché la mutazione (G>A) al codone 26 crea un sito di splicing alternativo all'interno del 1° esone. Di conseguenza gli eterozigoti AE, e gli omozigoti EE mostrano alcune caratteristiche talassemiche, ma di solito sono clinicamente asintomatici (1). I soggetti portatori di queste condizioni genetiche possono contribuire a produrre il composto Hb E/ β-talassemia se il partner è portatore di β-talassemia (Fig. a).
Il Paziente 2 è risultato portatore di un composto eterozigote [HBB:c.79G>A; β 26(B8) Glu>Lys (Hb E) allo stato eterozigote / HBB:c.92+5G>C; IVS-I-5 (G->C) β+ allo stato eterozigote]. La caratteristica saliente nel Paziente 2, come riportato in Tabella, è la presenza elevata di Hb F e l’anemia. L’Hb A rilevata (4,7%) è dovuta, in questo caso, al difetto β+-talassemico riscontrato e pertanto il Paziente 2 può avere figli malati che presentano un composto β-talassemico (β+/β0) con una probabilità del 25%, se la partner è portatrice di β0-talassemia e con la stessa probabilità avere figli con il composto Hb E/ β0-talassemia, clinicamente altrettanto importante (Fig.b).
L’Hb E/β-talassemia è una malattia comune in diverse parti del Sud-Est asiatico. Essa si traduce in un quadro clinico variabile, talvolta simile a quello della β-talassemia omozigote che varia da una condizione indistinguibile rispetto alla talassemia major (TDT) ad una forma lieve di talassemia intermedia (NTDT). Le condizioni più gravi si trovano in individui con Hb E e βo-talassemia, che di solito hanno circa 50-70% di Hb F. I livelli di emoglobina possono abbassarsi fino a 4-5 g/dl, conseguentemente la gestione clinica di questi pazienti è simile a quella dei soggetti con talassemia major. Gli eterozigoti composti per Hb E e β+-talassemia di solito presentano un fenotipo clinico più lieve e producono quantità variabili di Hb A in presenza di mutazioni di tipo β++, come nel caso dei difetti (più frequenti in Asia) -28 (A>G) e codone 19 (G>A). Alcuni pazienti con gravi mutazioni di tipo β+, come IVS I-5 (G>C) e IVS II-654 (C>T), possono produrre sintomi gravi come quelli tipici dell’Hb E/β0-talassemia. Analogamente a quanto avviene i alcuni casi di β-talassemia omozigote, i fattori genetici che spiegano un fenotipo «mild» sono dovuti, come già detto, a mutazioni lievi di tipo β++, ma anche alla co-ereditarietà della α-talassemia o alla presenza del sito di restrizione XmnI, prodotto dal polimorfismo C>T in posizione -158 in 5′ al gene della Gγ-globina, allo stato omozigote (2,3). Il composto eterozigote Hb E/β0-talassemia costituisce la forma grave più comune di talassemia nel Sud-Est asiatico ed in alcune parti del subcontinente indiano. Tuttavia, i recenti cambiamenti demografici hanno fatto sì che tale composto rappresenti un problema di salute anche in altre parti del mondo.
CONCLUSIONI
Il laboratorio, con gli esami di 1° livello e la conferma dell’Hb E mediante test alternativi, può concludere il suo percorso diagnostico disponendo delle informazioni sufficienti a distinguere genericamente l’Hb E omozigote dal composto Hb E/Beta Talassemia. La caratterizzazione molecolare per una diagnosi alla nascita può essere prevista nei casi di soggetti trasfusi, quando occorre definire con precisione il quadro dei difetti genetici per una gestione clinica o una consulenza genetica (4). In sintesi sappiamo che: L’Hb F è significativamente presente solo con Hb E/ β0-Talassemia o Hb E/ β+-Talassemia Fenotipi clinici intermedi o gravi si hanno solo con Hb E/ β0-Talassemia o Hb E/ β+-Talassemia L’anemia grave si osserva solo nei casi di Hb E/ β0-Talassemia I livelli di Hb A2 non consentono di discriminare tra Hb E omozigote e Hb E/ (β0 o β+ o β++)-Talassemia L’Hb A, nei soggetti non trasfusi, è presente solo nelle forme di Hb E/ β++ oppure di Hb E/ β+-Talassemia Alla nascita non è possibile distinguere l’Hb E omozigote dall’Hb E/ β0-Talassemia.
BIBLIOGRAFIA
1.Rees DC, Styles L, Vichinsky EP, Clegg JB, Weatherall DJ. The hemoglobin E syndromes. Ann N Y Acad Sci. 1998 Jun 30;850:334-43. doi: 10.1111/j.1749-6632.1998.tb10490.x. PMID: 9668555.
2.Hirsch RE, Sibmooh N, Fucharoen S, Friedman JM. HbE/β-Thalassemia and Oxidative Stress: The Key to Pathophysiological Mechanisms and Novel Therapeutics. Antioxid Redox Signal. 2017 May 10;26(14):794-813.
3.Fucharoen S, Weatherall DJ. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2012;2(8):a011734. Published 2012 Aug 1. doi:10.1101/cshperspect.a011734.
4.Barberio G, Ivaldi G. (2020). Emoglobinopatie. Dalla diagnosi alle consulenze specialistiche (Vol. 1). Piccin.
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *