INTERPRETAZIONI
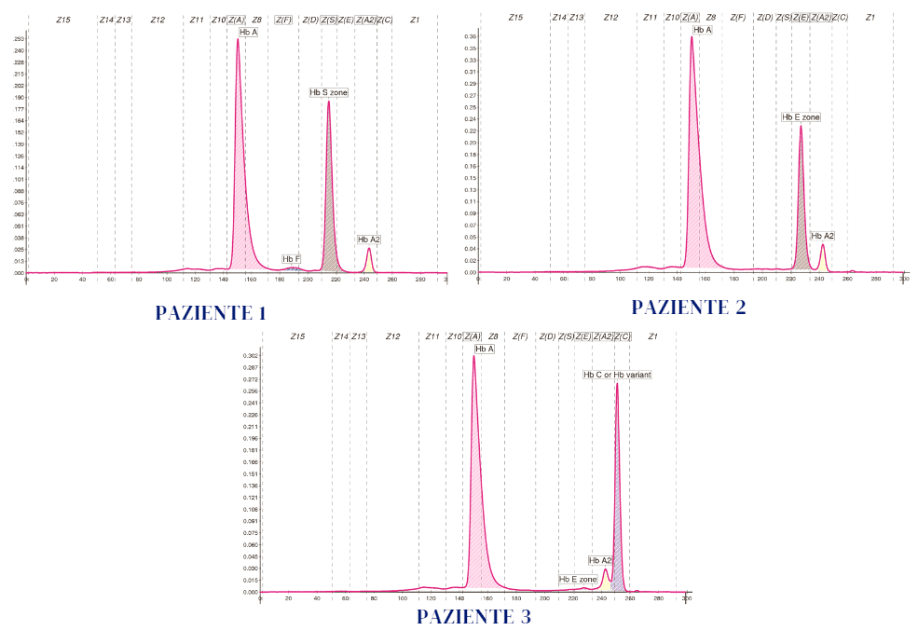
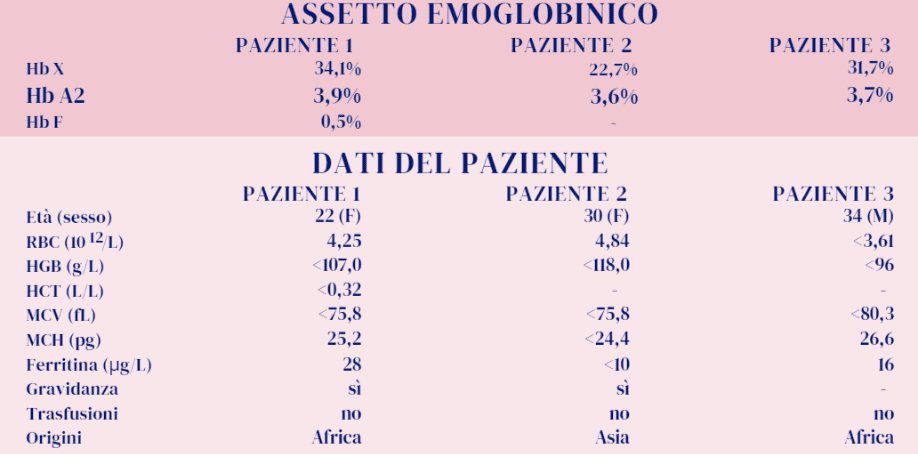
Obiettivo di questo «caso clinico» è l’interpretazione appropriata del valore dell’Hb A2 e di altri parametri, in presenza di una variante beta, per evitare erronee conclusioni diagnostiche. Vengono considerati i risultati degli esami eseguiti su tre soggetti: Paziente 1 (Hb S), Paziente 2 (Hb E) e Paziente 3 (Hb C) riportati in tabella.
Le varianti sono state confermate, come previsto dalle linee guida per le emoglobinopatie, con metodi alternativi (test di sickling o 2° metodo separativo) (1). Le alterazioni dei parametri emocromocitometrici riscontrate e riportate in Tabella dipendono in parte anche dallecondizioni cliniche dei soggetti. Tali alterazioni non condizionano comunque il ragionamento deduttivo, sviluppato nel tentativo di riuscire a prevedere una eventuale condizione genetica associata alle singole varianti emoglobiniche. Le tre β-varianti portate in esempio sono le più osservate al mondo e riscontrate anche nella popolazione italiana con una certa frequenza inseguito ai cambiamenti prodotti dai flussi migratori. Per interpretare al meglio il valore dell’Hb A2, degli indicatori eritrocitari e della quantità divariante presente occorre considerare che:
- Il valore relativo di Hb A2 è il parametro utilizzato per conoscere l’eventuale presenza di un tratto β-talassemico e, quindi, nella prassi di laboratorio rappresenta un riferimento fondamentale che consideriamo «normale», cioè non associato a β-talassemia, quando è inferiore a 3,2% (1).
- Esistono numerose cause, genetiche e no, che possono alterare parzialmente il valore di tale parametro (2).
- La ricerca della β-talassemia, anche associata ad altri difetti globinici, rappresenta sempre il principale obiettivo dei percorsi di prevenzione nella popolazione italiana (1).
- Nella normalità, i geni deputati alla sintesi di catene β-globiniche sono due, uno per ciascun cromosoma 11. Quando uno dei due alleli è «talassemico», il prodotto che deriva dall’unico gene β capace di tradurre l’informazione contribuisce a produrre la totalità o quasi (a seconda del tipo di mutazione) delle catene globiniche che potranno andare a formare il tetramero funzionante di emoglobina.
- Quando è presente, oltre ad un difetto qualitativo (es. Hb S), anche un difetto quantitativo (β-talassemia), l’emoglobina S avrà una percentuale compresa in genere tra il 50% e il 90%. Ciò vale per gran parte delle β-varianti co-ereditate con un difetto β-talassemico. La variabilità della percentuale relativa di composto Hb S/β-Tal dipende dall’estrema eterogeneità dei difetti strutturali e di quelli β-talassemici; questi ultimi saranno caratterizzati da una mancanza di sintesi delle catene β-normali (β0-Tal) o da una sintesi diversamente presente (β+-Tal, β++ -Tal, β+++ -Tal).
- Le β-varianti presentano generalmente una percentuale relativa che può dipendere anche dalla loro diversa affinità nei confronti delle catene α. Di solito tale affinità è minore rispetto a quella delle catene β normali. Anche nel caso di contemporanea presenza di a-talassemia, si riducono le percentuali relative sia della variante sia dell’Hb A2 (1).
- Nei composti eterozigoti formati da β-varianti con β-talassemie si ha quasi sempre la presenza di Hb F in quantità molto variabile (3).
- L’eventuale presenza di β-varianti instabili, comunque rare, deve essere sempre valutata.
CONCLUSIONI
Il laboratorio che esegue esami di 1° livello per le emoglobinopatie, se è in grado di confermare le β-varianti più frequenti con metodi alternativi, potrà commentare i risultati ottenuti (4,5) considerando che in un soggetto adulto non trasfuso:
- Una percentuale borderline o aumentata dell’Hb A2 (di solito non superiore a 4,0%) non è necessariamente prodotta dalla presenza di un tratto β-talassemico.
- Una microcitosi, anche importante, può essere dovuta a numerose cause (tra cui a-talassemia, carenza marziale, caratteristiche simil-talassemiche, instabilità delle varianti).
- Una percentuale della variante emoglobinica inferiore a 45-50% informa, nella maggior parte dei casi, che una variante β non è presente in associazione con il difetto β-talassemico.
- I tre pazienti qui considerati sono pertanto risultati rispettivamente portatori di Hb S, Hb E e Hb C e non è presente alcun difetto β-talassemico, pur presentando valori di Hb A2 al di sopra della norma .
BIBLIOGRAFIE
1.Raccomandazioni per la diagnostica di primo livello delle emoglobinopatie-SITE 2012. http://www.site-italia.org/collana scientifica.php. (ultimo accesso aprile 2019).
2.Ivaldi G, Barberio G, Harteveld C, et al. HbA2 measurements in beta-thalassemia and in other conditions. Thalassemia Rep 2014; 4:45–8.
3.Mosca A, Paleari R, Leone D, et al. The relevance of hemoglobin F measurement in the diagnosis of thalassemias and related hemoglobinopathies. Clin Biochem 2009;42(18)1797-801.
4.Ivaldi G, Barberio G, Carta M, et al. Diagnosi di laboratorio e prevenzione delle emoglobinopatie: considerazioni e proposte sulla comunicazione del risultato degli esami di primo livello. Biochim Clin 2010;34:277-82.
5.Barberio G, & Ivaldi G. (2020). Emoglobinopatie. Dalla diagnosi alle consulenze specialistiche (Vol. 1). Piccin.
Scrivi le tue impressioni e i commenti, verranno pubblicati il prima possibile!
Il tuo indirizzo email non sarà pubblicato. I campi obbligatori sono contrassegnati *